FDA’s Disruptive Final Rule: A Glimpse into its Impact on Laboratory Developed Test (“LDT”) Manufacturers
-
October 09, 2024
-

The U.S. Food and Drug Administration (“FDA”) has introduced a significant regulatory shift with its Final Rule, effective July 5, 2024, redefining the oversight of Laboratory Developed Tests (“LDTs”).1 This rule explicitly categorizes LDTs as in-vitro diagnostic products (“IVDs”), subjecting them to the same rigorous enforcement and regulations as other IVDs.2 By phasing out its historical general enforcement discretion for LDTs, the FDA aims to protect the public health, ensuring the safety and efficacy of IVDs while considering the essential factors such as patient access and reliance. However, this rule has sparked controversy among industry stakeholders, who have voiced their concerns through extensive comments.3 Additionally, the FDA is facing legal challenges, including a lawsuit from the American Clinical Laboratory Association (“ACLA”) which employed the recent overturn of Chevron deference by the U.S. Supreme Court, potentially curbing the authority of federal agencies.4 This article explores the potential impact of the Final Rule on the regulatory landscape for LDTs and its implications for manufacturers.
Understanding Laboratory Developed Tests (“LDTs”)
Laboratory Developed Tests (“LDTs”) are diagnostic tests intended for clinical use, and are developed, validated, and used exclusively by individual Clinical Laboratory Improvement Amendments (“CLIA”) -certified laboratories. Unlike commercially distributed tests, LDTs range from routine diagnostics like blood counts and drug tests to more complex molecular and genetic assessments for conditions such as cancer, heart disease, and rare and infectious diseases.
What Is The Phaseout Policy And how Is It Being Enforced?
Historically, the FDA exercised a broad enforcement discretion for most LDTs, considering the lower risk associated with them at the time of the Medical Device Amendments of 1976 (the MDA). However, with the advancement in technology and the growing reliance on LDTs for critical healthcare decisions, the associated risks have increased significantly. LDTs are now widely used by diverse populations and often guide crucial medical decisions. In response, the FDA is phasing out this discretion over four years, gradually imposing new regulatory and compliance expectations on clinical laboratories.
The transition will occur in five stages, each adding layers of regulatory requirements for LDTs. Figure 1 provides an overview of these expectations and some key elements to be considered when implementing them.
Figure 1: Key Elements in Implementing the Five Phaseout Stages of FDA’s Final Rule5
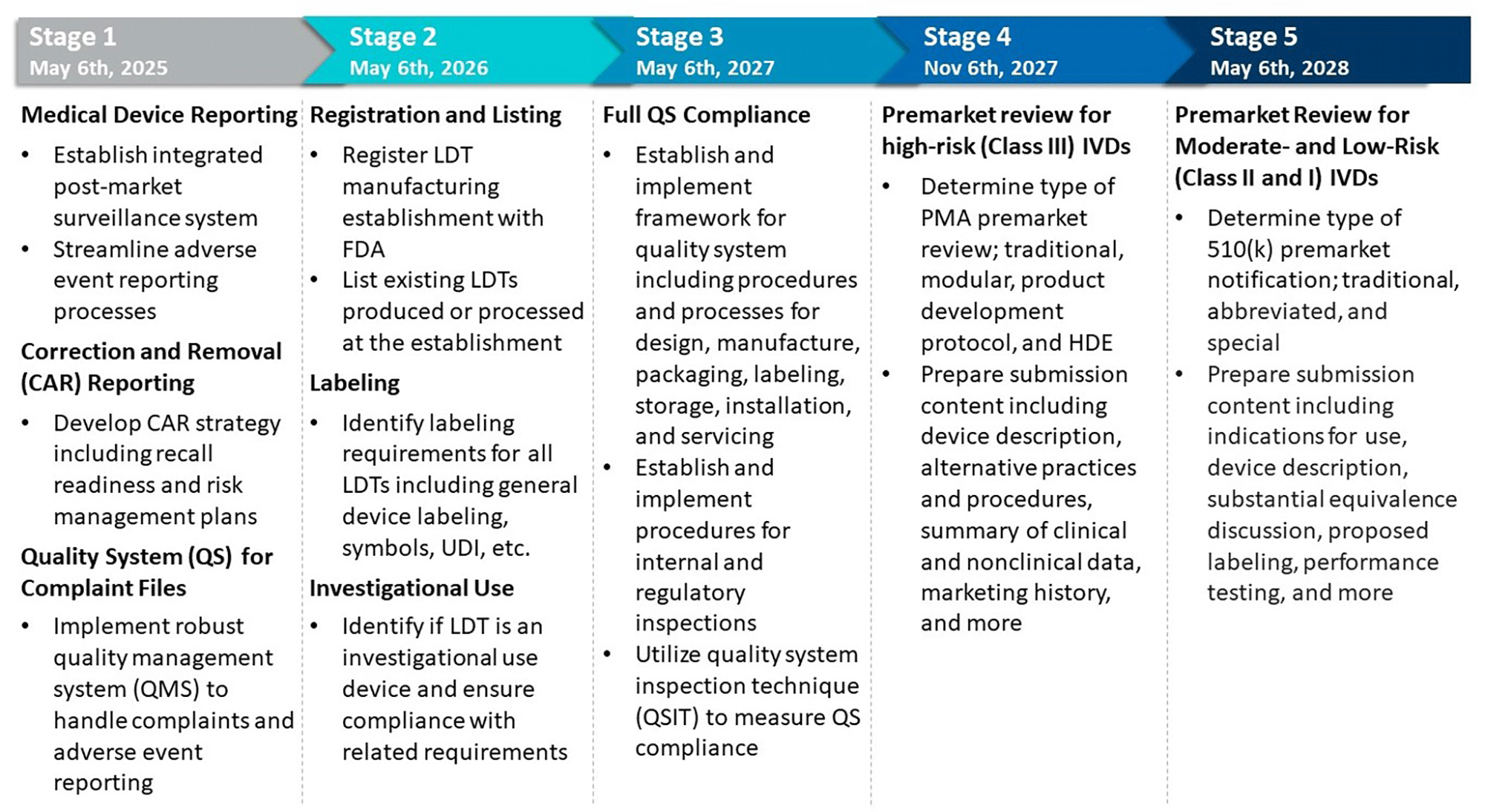
The Final Rule allows “targeted” enforcement discretion for specific categories of IVDs offered as LDTs, as outlined in Figure 2.
Figure 2: Enforcement Discretion from Applicable Stages of Phaseout Policy for Certain LDTs6
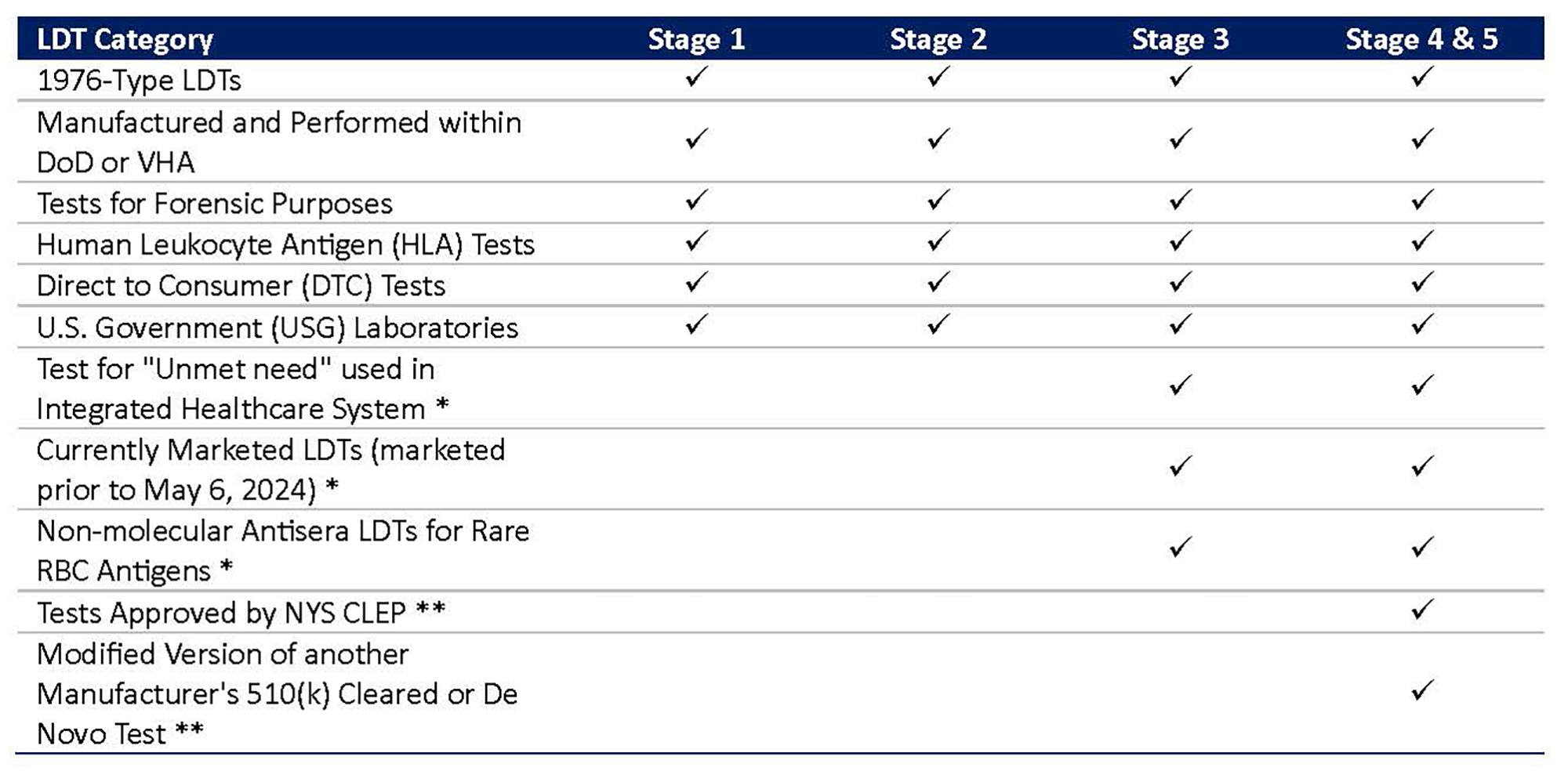
*FDA intends to exercise enforcement discretion and generally not enforce premarket review and QS requirements (except for requirements under Part 820, Subpart M (Records)).
** FDA generally will not expect compliance with 21 CFR Part 820 requirements other than design controls, purchasing controls, acceptance activities, CAPA, and records requirements.
New Regulatory Pathway for LDTs
Following the Final Rule, all IVDs offered as LDTs will be subject to the same stringent regulatory pathway as traditional IVDs. This pathway applies not only to new LDTs but also to existing ones undergoing significant modifications.
Figure 3: New FDA Approval Process for LDTs
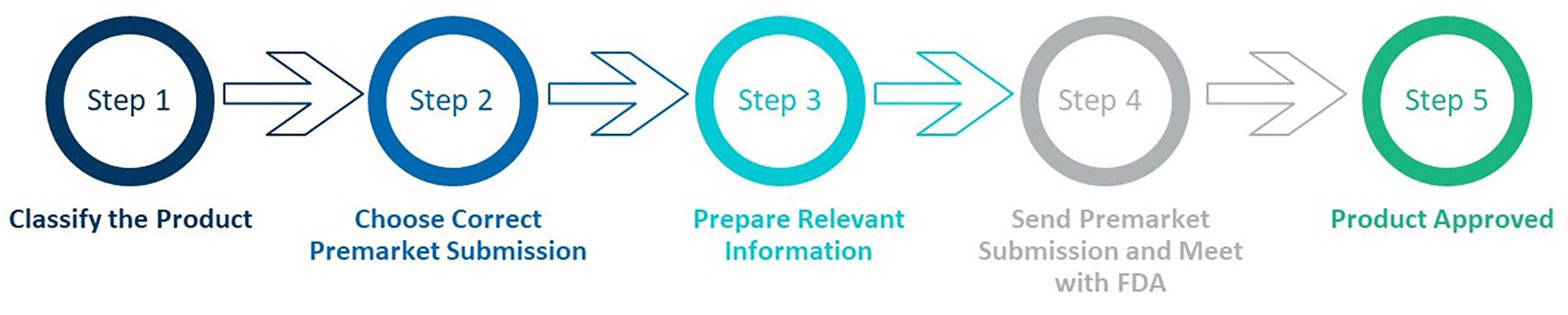
LDT manufacturers will be required to follow a new regulatory approval process, as outlined in Figure 3. This process can be broken down into five steps:
- Classification: Determine if the LDT is Class I (low to moderate risk), Class II (moderate to high risk), or Class III (high risk).
- Pre-market Submission Selection: Choose the correct submission type (510(k), PMA, De Novo, and HDE (Humanitarian Device Exemption)).
- Preparation of Relevant Information: Conduct pre-submission meeting with FDA, if necessary, and compile necessary data, including design controls, non-clinical testing, clinical evidence, and labeling.
- Premarket Submission: Submit to the FDA, pay associated user fees, complete establishment registration and product listing, and meet with the FDA if required.
- Product Approval: Follow post-market requirements, including product tracking and reporting malfunctions, serious injuries, or deaths.
This new regulatory process represents a significant departure from the previous LDT approval pathway, imposing substantial regulatory and financial burden on manufacturers.
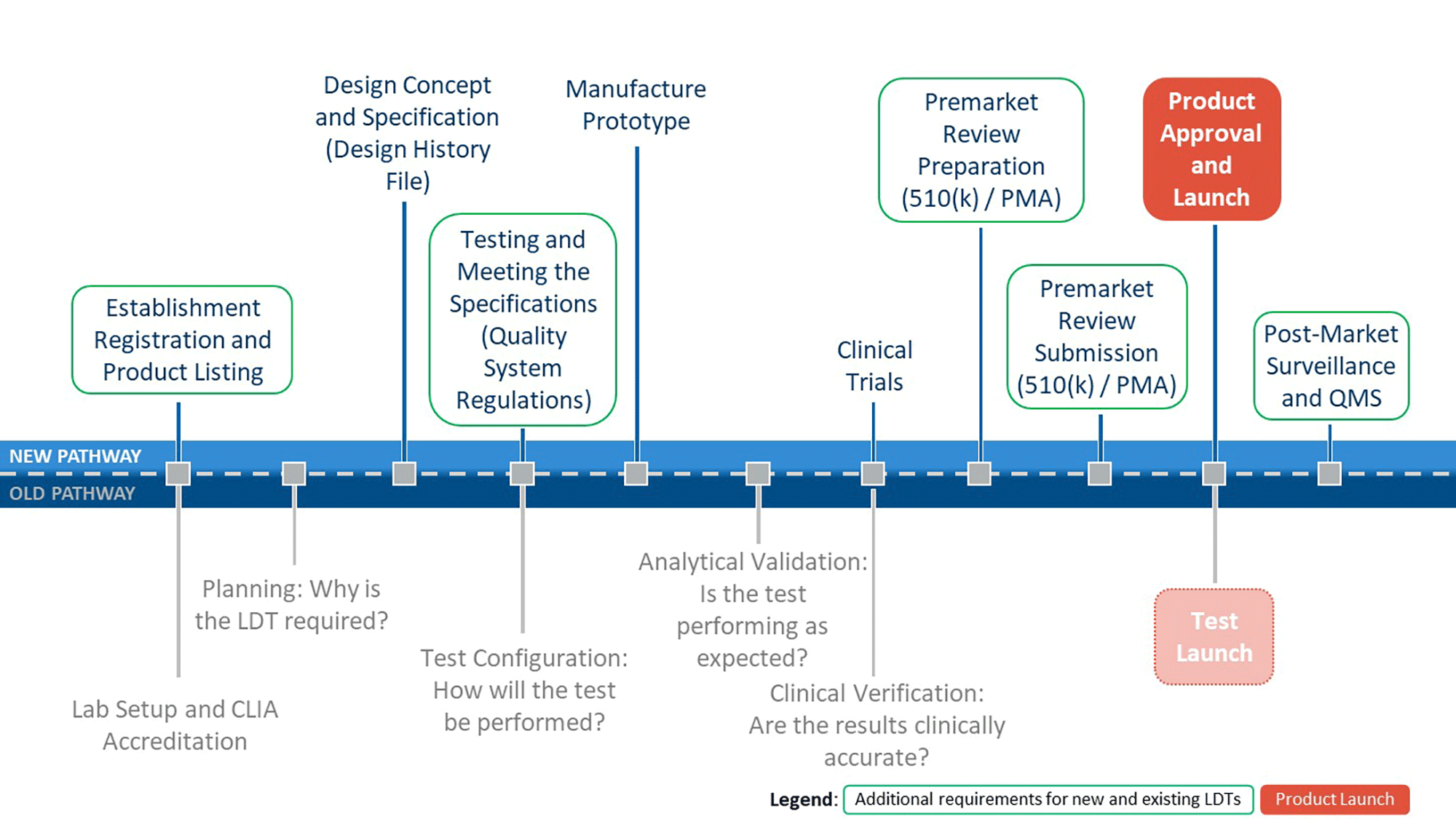
Figure 4 provides an overview of some additional requirements that LDT manufacturers would be required to meet under this Final Rule.
Figure 4: Comparison of Old and New Regulatory Pathways for LDTs
Here’s How the Final Rule Will Impact Each Stakeholders
The regulation of LDTs has been an old topic of debate between the FDA and the industry. The FDA has received over 6,500 comments on its initial draft.7 The Final Rule will have huge implications on the medical product development and manufacturing, on clinical laboratories involved in LDTs development, patients, as well as suppliers and third parties involved in LDT development.
Potential Impact to LDT Manufacturers: There will be an anticipated, unwanted effect of the Final Rule on 2,362 existing and 189 new laboratories that manufacture LDTs, with the highest impact on small laboratories.8 Key changes include:
- More stringent pre-market review requirements
- Increased cost associated with premarket submissions, QS and annual reporting requirements
- Enhanced scrutiny of quality standards and clinical validation
- Delays in market introduction and increased time-to-market for new LDTs
- Innovation slowdown due to the additional regulatory hurdles
Potential Impact to Patients: Users of LDTs will mostly benefit from the Final Rule; however, there may be some unwanted consequences on certain patient groups. The patients will have:
- Enhanced safety and access to more accurate and reliable tests
- Increased confidence on medical recommendations based on test results
- Issues related to access, information and limited availability of certain tests due to being less affordable or accessible
Potential Impact to Third Parties: Suppliers who manufacture parts and accessories for LDTs will also see an impact of the Final Rule on their daily operations. This includes:
- Adjustments to manufacturing processes to comply with the new regulations
- New market opportunities and growth for providers of FDA-compliant components
- Higher demand for regulatory consultants and legal services
- Increased costs for providers of quality control and compliance
Navigating The New Regulatory Landscape: Strategies for the Next Four Years
As the FDA phases in the Final Rule over the next four years, manufacturers must take proactive steps to navigate the complexities of the new regulatory pathways for LDTs. With the effective date for Stage 1 approaching on May 6, 2025, it is crucial for laboratories to fully understand the extensive requirements that will govern their operations. This includes:
- Establishment Registration and Device Listing: Identifying FDA classifications, preparing and submitting regulatory applications, and completing establishment and product registration.
- Compliance with Quality System Regulations: Developing and implementing comprehensive quality systems, including design and process controls, documentation and corrective actions.
- Premarket Review and Submission: Providing guidance and support through the premarket review process until FDA approval is obtained.
- Post-Market Surveillance and QMS: Facilitating the adoption post-market surveillance processes, enhancing organizational awareness, and implementing quality management systems.
While the goal of enhancing patient safety and ensuring the efficacy of diagnostic tests is commendable, the burden of compliance—especially for smaller laboratories—is significant.
Footnotes:
1: “Medical Devices; Laboratory Developed Tests”, Federal Registrar, (06 May 2024).
2: “ACLA Challenges FDA’s Final Rule to Regulate Laboratory Developed Testing Services as Medical Devices”, American Clinical Laboratory Association, (29 May 2024).
3: Ibid.
4: Ibid.
5: Ibid.
6: Ibid.
7: “Overview of FDA Laboratory Developed Test Final Rule”, The American Society for Clinical Pathology, (14 May 2024).
8: Ibid.
Published
October 09, 2024
 Key Contacts
Key Contacts
Senior Consultant