The Medical Device Regulation (“MDR”)
Need-to-Know Changes
-
June 20, 2022
Downloads Download Article
Download Article
-

The Medical Device Regulation (“MDR”) is a new set of regulations that govern the manufacturing and distribution of medical devices in Europe, and replaced the Medical Devices Directive (“MDD”), which was effective until May 2021. Ensuring compliance with MDR is mandatory for all medical device companies that operate in the European market, and transition to the new regime is required by May 2024. This change in regulation regime has considerable impacts on the pharmaceutical industry, as this short summary reveals.
The MDD was intended to harmonise laws relating to medical devices within the European Union (“EU”). For a manufacturer to legally place a medical device in the European market, the requirements of MDD must be met.
On 26 May 2021, MDD was replaced by MDR, which was intended as an improved version of the regulation. This provides consistency in the standards for quality and safety measures across all 27 EU member states (excluding the UK).
The MDR document is significantly longer and more rigorous than the original MDD. A strong emphasis has been placed on product safety, hygiene and postmarket surveillance. This has been triggered by the increasing prevalence of medical device malfunction, which has in turn prompted an increased demand for the transparency of technical information.
The new rules require companies to review their products and core processes. This includes recertifying existing products as well as updating technical documentation and labelling to ensure that the new standards are met.
Whilst MDR is not completely different from MDD, companies will need to make a significant number of changes to technical documentation for products and existing processes in their quality management systems (“QMSs”) to ensure that they comply. The new standards are also stricter and more evidence based.
Some key changes as a result of MDR:
Equivalence
This will be more thoroughly
interpreted, making it more
challenging to demonstrate
clinical safety or performance of
medical devices.
Unique device identification
(“UDI”)
This will be implemented to help
track devices throughout the
economic operator supply chain
and will be required on all labels.
Definition of medical device
This will be broadened to include
non-medical and cosmetic devices
that were not previously regulated.
In terms of how these will tangibly impact medical device companies:
- Companies undergoing the transition will need to revisit core processes, including quality assurance and risk management, as well as postmarket expectations.
- Balancing business as usual with the transition to MDR may prove challenging for businesses from a resourcing perspective. The transition to MDR requires careful review, planning and updating of processes and procedures for compliance whilst still maintaining business as usual
- Companies will face significant time pressures, on 26 May 2024, all MDD certificates will become void. This makes it imperative that companies act now.
What are the life cycle activities directly impacted by MDR?
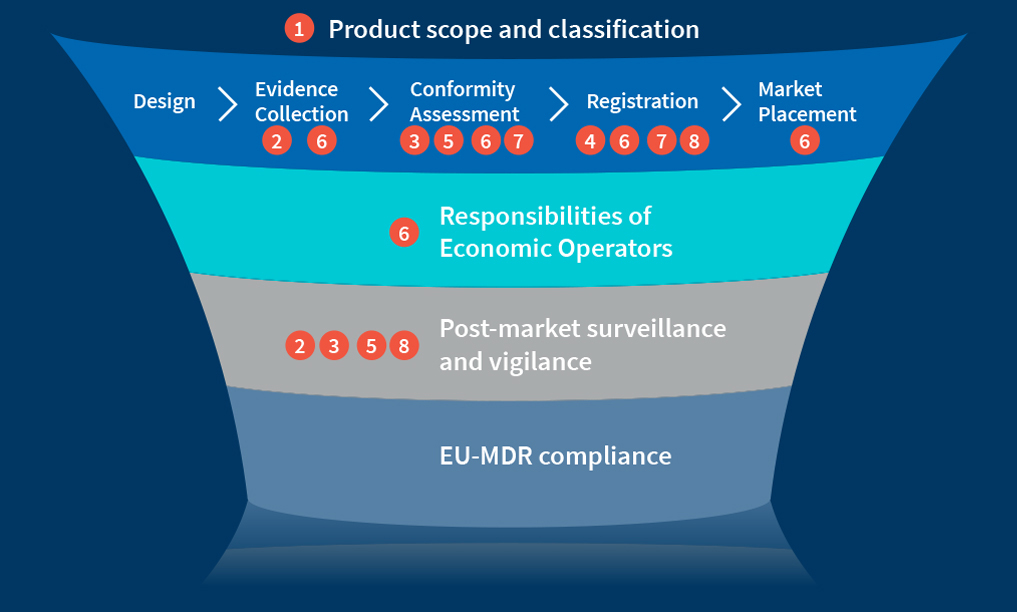
Figure 1: EU-MDR compliance
The new regulations seek to increase medical device safety and effectiveness in the European market while addressing the weaknesses that were revealed in the implementation of MDD by several medical device manufacturers.
The regulations feature several significant changes, including those in the following areas:
- Scope and classification of products
- Clinical evaluation and investigations
- Safety and performance requirements
- Product traceability through UDI/implant card
- Post-market surveillance and vigilance
- Role of economic operators and person responsible for regulatory compliance
- Role and scope of notified body safety and clinical performance recording and reporting
Medical device manufacturers must consider the following areas in light of MDR to ensure compliance by May 2024 and effectiveness going forward:
Ensure a compliant MDR transition
- Carry out a review of technical documents to ensure that they comply with EU MDR requirements.
- Reassess clinical data for devices that are already in the market.
- Conduct additional testing for recertification for products with insufficient data.
Conduct a critical review of QMSs
- Establish strategies to ensure regulatory compliance.
- Introduce procedures to perform clinical evaluations, identify safety requirements and manage risk.
- Maintain vigilance and handle communications with relevant authorities.
- Implement processes to make sure quality standards are upheld, ensuring maintenance, implementation and upkeep of the QMS.
- Set up and manage a post-market surveillance programme (“PMS”) and create a process to analyse and act on the information collected.
Establish post-market surveillance
- Continually reassess risk by collecting information on device performance and how it compares to similar products in the market and threshold values.
- Implement processes to assess information and enable manufacturers to implement corrective actions.
- Communicate with relevant parties and notified bodies.
- Introduce tools to trace and identify devices that require corrective actions.
FTI Consulting can help support companies with these new challenges
Our team has completed numerous projects across the pharmaceutical and life sciences sectors, and are able to leverage our previous experience to support transformation programmes.
We have proven experience in helping medical device companies take control of their MDR programme, rapidly resolve complex issues and ensure they comply with the necessary regulatory requirements.
For more information or to discuss how these changes impact you and your organisation, please get in touch at BusinessTransformationEMEA@fticonsulting.com or contact one of the team members below.
Published
June 20, 2022
 Key Contacts
Key Contacts
Managing Director